Griscelli syndrome
Griscelli syndrome
Natural Standard Monograph, Copyright © 2013 (www.naturalstandard.com). Commercial distribution prohibited. This monograph is intended for informational purposes only, and should not be interpreted as specific medical advice. You should consult with a qualified healthcare provider before making decisions about therapies and/or health conditions.
Related Terms
Albinism, Griscelli disease, Griscelli syndrome type 1, Griscelli syndrome type 2, Griscelli syndrome type 3, Griscelli syndrome with neurologic impairment, Griscelli syndrome-cutaneous and neurological type, Griscelli-Prunieras syndrome, Griscelli-Prunieras variant, Griscelli's disease, GS, GS1, GS2, GS3, partial albinism, partial albinism and primary neurologic disease without hemophagocytic syndrome, partial albinism with immunodeficiency.
Background
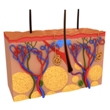
Griscelli syndrome (GS) is a rare genetic disorder that primarily affects the skin and hair. GS belongs to a group of conditions known as albinism and is named after Claude Griscelli, who first described this syndrome. This group of conditions is marked by decreased or absent pigment (coloring) in the skin, hair, and eyes. In some cases, albinism causes eye problems, including decreased pigment in the iris.
Albinism is caused by mutations in genes that encode for making the pigment melanin, which provides color to hair, skin, and eyes. Melanin also absorbs ultraviolet light from the sun in order to protect the skin. Because melanin is absent or decreased in GS, affected individuals are at increased risk of skin damage caused by the sun, including skin cancer.
People with GS tend to have a milder form of albinism characterized by pale skin and silvery gray hair at birth. In addition, they tend to develop clumps of pigment at the hairline and in melanocytes, a type of skin cell. Individuals with GS may also be prone to developing neurological and immune system disorders. There are several types of GS, which include Griscelli syndrome types 1, 2, and 3.
GS is caused by mutations in one of three genes. These are the RAB27A,MYO5A, and MLPH genes, which provide instructions for the proper production and transport of melanin.
GS is inherited, or passed down among family members, as an autosomal recessive trait. Individuals receive two copies of most genes, one from the mother and one from the father. To inherit an autosomal recessive trait, an individual must inherit two recessive copies of the causative gene.
GS is extremely rare, and its exact prevalence is unknown. There have been about 60 cases reported worldwide and fewer than 10 reported in the scientific literature in the United States. Internationally, GS has been reported in people of Indian, Turkish, and other Mediterranean descent. GS appears to affect males and females in equal numbers.
The prognosis for those with GS is generally poor. Without bone marrow transplantation, individuals with this syndrome tend to die by five years of age.
Risk Factors
Griscelli syndrome (GS) is inherited, or passed down through family members. Therefore, the only known risk factor is a family history of the disorder. GS is extremely rare, and its exact prevalence is unknown. There have been about 60 cases reported worldwide and fewer than 10 reported in the United States. Internationally, GS has been reported in people of Indian, Turkish, and other Mediterranean descent. GS appears to affect males and females in equal numbers.
Because GS is inherited as an autosomal recessive trait, an individual needs to inherit two recessive copies of the causative gene for the disease to occur. Individuals who have only one defective copy of the causative gene have mild or no symptoms and are called carriers, because they may pass on the disorder to their children. If one parent is a carrier, then each child has a 50% chance of inheriting one defective gene and also being a carrier. If both parents are carriers, each child has a 25% chance of inheriting two defective genes, a 50% chance of inheriting only one defective gene, and a 25% chance of inheriting neither defective gene. Therefore, if both parents are carriers, about one out of four children may have the disorder.
Causes
Genetic mutations: People with Griscelli syndrome (GS) tend to have a milder form of albinism characterized by pale skin and silvery gray hair at birth. In addition, they tend to develop clumps of pigment at the hairline and in melanocytes, a type of skin cell. Individuals with GS may also be prone to developing neurological and immune system disorders. Albinism is caused by mutations in genes that provide instructions for making the pigment melanin, which provides color to hair, skin, and eyes. Melanin also absorbs ultraviolet light from the sun to protect the skin. Because melanin is absent or decreased in GS, affected individuals are at increased risk of skin damage caused by the sun.
GS is caused by mutations in one of three genes. These are the RAB27A gene, which provides instructions for making the GTP-binding protein Rab27a; the MYO5A gene, which provides instructions for making the myosin motor protein myosin5a; and the MLPH gene, which provides instructions for making the melanophilin protein. These proteins are important for the proper production and transport of melanin. Rab27a and Myo5a proteins are responsible for the movement of organelles (vesicles) within the cell that carry melanin. Defects in these genes result in pigment dilution. Defects in the MYO5A gene cause neurologic pathology, because this gene is expressed in the central nervous system, mainly the brain, whereas defects in the RAB27A gene do not cause neurologic defects, because this gene is not found in the brain. The MLPH gene encodes for melanophilin, which is involved in melanosome transport and serves as a link between melanosome-bound RAB27A and the motor protein MYO5A. Together, these defects decrease the transport of melanin.
Autosomal recessive inheritance: GS is inherited, or passed down among family members, as an autosomal recessive trait. Individuals receive two copies of most genes, one from the mother and one from the father. To inherit an autosomal recessive trait, an individual must inherit two defective copies of the causative gene.
Individuals who have only one defective copy of the causative gene have mild or no symptoms and are called carriers, because they may pass on the disorder to their children. If one parent is a carrier, then each child has a 50% chance of inheriting one defective gene and also being a carrier. If both parents are carriers, each child has a 25% chance of inheriting two defective genes, a 50% chance of inheriting only one defective gene, and a 25% chance of inheriting neither defective gene. Therefore, if both parents are carriers, about one out of four children will have the disorder.
There is a lack of evidence to suggest that GS can occur through a spontaneous mutation.
Signs and Symptoms
General: Signs and symptoms of Griscelli syndrome (GS) tend to emerge between the ages of four months and four years. Individuals who have GS caused by mutations in the MYO5A gene (type 1) tend to show symptoms earlier than those who have GS caused by mutations in the RAB27A gene (type 2). Individuals with a mutation in the MLPH gene (type 3) do not present with any immune or neurological symptoms; this type is characterized by decreased melanin.
Cognitive problems: Some individuals with GS may have cognitive disabilities. The severity of these disabilities and the proportion of people affected are not known.
Eye dysfunction: Some individuals with GS may have decreased function of the retina, a layer of cells in the back of the eye that send information to the brain via the optic nerve. Decreased function of the retina may lead to blindness. Some individuals with GS may have decreased pigment in the iris, which can lead to increased sensitivity to light.
Hair problems: Often the first visible sign of the condition, people with GS tend to have silver hair. Other hair colors seen in people with GS include gray and grayish gold. In addition, people with GS tend to have clumps of pigment (coloring) at the hairline.
Immune system problems: Individuals may have decreased immune function that is apparent in the occurrence of severe viral and bacterial infections. These opportunistic infections are generally related to uncontrolled activity of lymphocytes, which are white blood cells important in defending the body against infection; and macrophages, which perform a similar function. The lymph nodes in the groin, neck, chest, and abdomen may become swollen.
Neurologic problems: Neurologic problems associated with GS tend to appear early in life. These include hydrocephaly (excessive accumulation of fluid in the brain), abnormal muscle tone, poor coordination, hemiplegia or hemiparesis (paralysis on one side of the body), seizures, delayed attainment or regression of developmental milestones, spasticity (persistent contraction of certain muscles), nystagmus (involuntary eye movements), facial paralysis, polio, and other brain problems or infections.
Skin abnormalities: People with GS tend to have skin tone that is lighter than normal but darker than that of others with more severe forms of albinism. They may also have skin lesions and abnormal deposits of melanin in melanocytes, a type of skin cell.
Other: Certain organs, such as the liver and spleen, may be enlarged in individuals with GS.
Types of the Disease
Griscelli syndrome type 1: Griscelli syndrome type 1 (GS1) is also known as Griscelli syndrome with primary neurologic impairment and without immunologic impairment. This form of the disease is caused by mutation in the MYO5A gene, which encodes for making the myosin5a (MYO5A) protein (also known as the myosinVA or MYOVA protein). The MYO5A protein is responsible for the movement of organelles (vesicles) within the cell that carry the pigment melanin. Defects in this gene result in pigment dilution. The neurologic effects of GS caused by defects in the MYO5A gene usually appear very early in life. Severe neurologic manifestations in GS are associated with defects in the MYO5A gene. Severe neurologic symptoms are noticeable at birth, without any sign of an accelerated phase or regression over time. Symptoms include seizures, slow development of motor function, absence of coordinated voluntary movements, hypotonia (low muscle tone), and various structural abnormalities. Infections are not present in individuals with a MYO5A defect.
Griscelli syndrome type 2: Griscelli syndrome type 2 (GS2) is also known as Griscelli syndrome with immune impairment. This form of the disease is caused by mutations in the RAB27A gene, which provides instructions for making the GTP-binding protein RAB27A. The RAB27A protein is responsible for the movement of organelles (tiny organs) within the cell that carry the pigment melanin. Defects in this gene result in pigment dilution. GS caused by the RAB27A mutation can also cause neurologic manifestations in association with GS in the accelerated phase. Neurologic problems may be the first sign of GS in the accelerated phase. Neurologic manifestations occurring in patients with GS caused by the RAB27A mutation may not be as severe as those found in GS caused by MYO5A mutations. Symptoms include seizures, hypotonia, and intracranial hypertension, which is characterized by vomiting and altered states of consciousness. A history of severe infections associated with uncontrolled lymphocyte and macrophage activation can be present in patients with mutations in the RAB27A gene. GS2 typically progresses rapidly and may cause death by 1-4 years of age.
Griscelli syndrome type 3: Griscelli syndrome type 3 (GS3) is characterized by decreased melanin but no immune system or neurologic problems. This form of the disease is caused by mutations in the melanophilin gene (also known as MLPH), which provides instructions for making the melanophilin protein. This protein serves as a link between melanosome-bound RAB27A and the motor protein MYO5A. Together, these defects decrease the transport of melanin. Some researchers suggest that GS3 may also be caused by certain mutations in the MYO5A gene.
Diagnosis
General: Griscelli syndrome (GS) symptoms usually emerge from four months to four years of age. The first observed sign of GS is often silver hair, which indicates decreased production of the pigment melanin, which provides color to hair, skin, and eyes. Neurological symptoms are usually next to develop and may even be present at birth.
Biopsy: In a biopsy, a small sample of tissue is taken and evaluated in a laboratory. People suspected of having GS may undergo biopsies of the skin. A skin biopsy may be examined using a transmission electron microscope, which can also help distinguish GS from other types of albinism. Depending on individual patient characteristics, biopsies may also be performed on the liver and spleen to test for inflammation, which may occur in individuals with GS.
Blood tests: Blood samples may be taken to evaluate the white blood cell and platelet numbers and function, both of which may be decreased in individuals with GS; liver function, which may be decreased in patients with GS; and protein in the blood, which may be related to the bone marrow dysfunction in individuals with GS.
Genetic testing: DNA testing may be performed to confirm a diagnosis of GS. A sample of the patient's blood is taken and analyzed in a laboratory for defects in the RAB27A,MYO5A, and MLPH genes. If defects are detected, a positive diagnosis is made.
Hair analysis: A hair bulb tyrosinase assay can help distinguish GS from other types of albinism. In this assay, hair bulbs or roots from the scalp are plucked and placed in a solution containing dihydroxyphenylalanine (L-DOPA). In people with oculocutaneous albinism type 1, hair bulbs remain white in this solution, while those from people with other forms of albinism (including GS) turn dark. Examination of hair shafts using a polarized light microscope may help distinguish GS from other types of albinism.
Imaging studies: Ultrasound, a noninvasive procedure that uses sound waves to create a moving image of internal structures, may be used to assess the size and function of the liver and spleen, as these organs may be enlarged by inflammation in individuals with GS.
Physical exam: GS may be suspected based on the observations of decreased pigment (coloring) in the hair and skin. Individuals suspected of having GS should receive a thorough physical exam and should provide a complete family history. In addition, the patient or caregiver should be asked specific questions regarding the patient's development and history of infections.
Prenatal DNA testing: If there is a family history of GS, prenatal testing may be performed to determine whether the fetus has the disorder. Amniocentesis and chorionic villus sampling (CVS) can diagnose GS. However, because there are serious risks associated with these tests, patients should discuss the potential health benefits and risks with a medical professional.
During amniocentesis, a long, thin needle is inserted through the abdominal wall and into the uterus, and a small amount of amniotic fluid is removed from the sac surrounding the fetus. Cells in the fluid are then analyzed for normal and abnormal chromosomes. This test is performed after 15 weeks of pregnancy. The risk of miscarriage is about one in 200-400 patients. Some patients may experience minor complications, such as cramping, leaking fluid, or irritation where the needle was inserted.
During CVS, a small piece of tissue (chorionic villi) is removed from the placenta between the ninth and 14th weeks of pregnancy. CVS may be performed through the cervix or through the abdomen. The cells in the tissue sample are then analyzed for the mutated gene. Miscarriage occurs in about 0.5-1% of women who undergo this procedure.
Complications
Developmental delays: Some individuals affected with Griscelli syndrome (GS) may have delays in achieving early developmental milestones, such as gross and fine motor skills and expressive and receptive language; some may also have intellectual disabilities. Severe intellectual disability, characterized by an intelligence quotient (IQ) of under 70, is very rare. The presence of these developmental disabilities may require specialized programs in school to address specific disabilities.
Infections: People with GS caused by mutations in the RAB27A gene are prone to serious viral and bacterial infections.
Neurologic: Some people with GS may experience seizures. Patients with GS may also have hydrocephaly (an excessive accumulation of fluid in the brain), which can cause headaches, vomiting, nausea, sleepiness, or coma; poor coordination, which can result in falls; and spasticity (persistent contraction of certain muscles), which can lead to muscle fatigue.
Skin: People with any form of albinism, including GS, lack the pigment melanin, a material that changes the color of light it reflects and protects the skin from the harmful ultraviolet rays of the sun. This lack of pigment places people with GS at higher risk of sunburn, even if sun exposure is brief. Repeated exposure to the sun over the course of several years may cause skin to become rough and thick (called pachydermia) and may cause the development of precancerous and cancerous lesions. Because melanin is absent or decreased in GS, affected individuals are at increased risk of skin damage caused by the sun, including different types of skin cancer, such as basal cell and squamous cell carcinomas.
Treatment
General: There is no cure for Griscelli syndrome (GS). Instead, treatment aims to reduce symptoms and prevent complications. Depending on individual symptoms, individuals with GS should be regularly monitored by a geneticist, hematologist (blood specialist), dermatologist (skin doctor), neurologist (nervous system specialist), and pediatrician. Without bone marrow transplantation, GS results in death. The mean age at the time of death in people with GS is five years of age.
Activity restriction: Based on the presence of immunologic or neurologic problems, individuals with GS often limit participation in physical activities.
Bone marrow transplantation: Bone marrow transplantation, a procedure that transplants healthy bone marrow into a patient whose bone marrow is not functioning properly, may be a viable option for those with GS who are prone to frequent or severe infections. However, this procedure carries a number of risks, including rejection of the transplant. Immunosuppressants, medications that decrease immune function, may be prescribed to decrease the risk of rejection. Chemotherapy is given before the transplant and may cause diarrhea liver and lung damage. While waiting for the transplanted bone marrow to grow, a patient is still at risk for increased infection.
Corrective lenses: Eyeglasses are limited in their ability to improve vision in those with GS whose vision is impaired. Tinted lenses can help with sensitivity to bright light.
Medications: Antibiotics may be prescribed to those susceptible to frequent or severe bacterial infections. Side effects of antibiotics include diarrhea and upset stomach and may also include vomiting, white patches on the tongue. and a vaginal discharge. Although antibiotics are generally effective, overprescription of antibiotics can lead to drug-resistant bacteria that may not be treatable. Antiviral agents may be prescribed to those susceptible to frequent or severe viral infections. Some side effects of antiviral drugs may include headache, nausea, vomiting, loss of appetite, or insomnia. The effectiveness of antiviral drugs may depend on the targeted virus. Some viruses, such as the flu virus, that cause illness at one time may change, and therefore a new drug would need to be developed.
Anticonvulsant medications may help reduce the frequency and severity of seizures. Commonly prescribed anticonvulsants include topiramate (Topamax®), levetiracetam (Keppra®), zonisamide (Zonegran®), phenytoin (Dilantin®), carbamazepine (Tegretol®, Carbatrol®), phenobarbital, and valproic acid (Depakene®, Depakote®). The anticonvulsants are generally safe but in rare cases can cause serious side effects. Any of these medications can cause abnormalities in blood or platelet counts, as well as abnormalities in liver function, in addition to nausea, vomiting, and constipation. These drugs are generally accepted as effective for the treatment of seizures, although some medications work better for certain types of seizures.
Antineoplastic agents such as etoposide (VePesid®, Toposar®) may be used to treat the immune function problems common in GS. Immunosuppressants are medications that decrease the function of the immune system. These medications have been used with some success in treating individuals with GS. Some immunosuppressants include cyclosporine (Sandimmune®, Neoral®) and prednisone (Orasone®, Meticorten®, Sterapred®, Deltasone®). Immunosuppressive antibodies, such as antithymocyte globulins, are used with other immunosuppressive drugs and chemotherapy to treat the immune system problems distinctive of GS. Antimetabolites such as cytarabine (Cytosar-U®) and intrathecal methotrexate (Folex PFS®, Rheumatrex®) may be used to treat the immune system problems seen in GS.
All of these medications have several potential interactions and adverse effects. Their use should be thoroughly discussed with and monitored by a qualified healthcare professional.
Occupational therapy: Patients with GS may benefit from occupational therapy. During sessions, a therapist helps the child learn skills to help him or her perform basic daily tasks, such as eating, dressing, and communicating with others. Some patients work with therapists who specialize in disorders and disabilities. Parents and caregivers can ask their child's pediatrician for recommended therapists.
Sunscreen: There is no treatment for lack of pigment in people with albinism. However, because they are at increased risk for sunburn and skin cancer, people with albinism should wear a broad-spectrum sunscreen on all exposed areas of the body, including the scalp. Special clothing that increases protection from the sun is also available and includes hats, visors, long-sleeve shirts, and long pants.
Integrative Therapies
Note: Currently there is limited scientific evidence on the use of integrative therapies for the treatment or prevention of Griscelli syndrome (GS). The therapies listed below have been studied for related conditions such as sunburn and skin cancer. The integrative therapies listed below should be used only under the supervision of a qualified healthcare provider and should not replace other proven therapies.
Unclear or conflicting scientific evidence:
Chlorella: Early studies suggest a potential effect of chlorella on skin cancer. Avoid with known allergy or hypersensitivity to chlorella, its constituents, or members of the Oocystaceae family. Some children have been found to be allergic to chlorella. Chlorella has a high vitamin K content and may decrease the effectiveness of anticoagulants (blood thinners) such as warfarin. Long-term consumption of chlorella may cause manganese-induced parkinsonism. Other adverse effects include photosensitivity, occupational asthma, and fatigue.
Green tea: There is limited animal and human research on green tea as a protective agent from ultraviolet light injury to the skin. Some studies have found conflicting results. Comparisons have not been made with well-established forms of sun protection such as ultraviolet-protective sunscreen. The effects of green tea on skin damage caused by the sun remain unclear. Avoid if allergic or hypersensitive to caffeine or tannin. Use cautiously with diabetes or liver disease.
Lutein: Numerous laboratory studies have shown the antioxidant effect of lutein. More research is required on the use of lutein for sunburn prevention before a firm conclusion can be made. Avoid if allergic or hypersensitive to lutein or zeaxanthin. Use cautiously if at risk for cardiovascular disease or cancer. Avoid if pregnant or breastfeeding.
Lycopene: Lycopene, in combination with other carotenoids, such as beta-carotene, vitamins C and E, selenium, and proanthocyanidins, may help in reducing sunburn. Selected protective effects from ultraviolet (UV) rays have been observed in small, short-term studies. More research is needed before a firm conclusion can be drawn. Avoid if allergic to tomatoes or to lycopene. Avoid if pregnant or breastfeeding.
Para-aminobenzoic acid (PABA): PABA is best known for its topical use as a component of sunscreen products. Although PABA and related compounds have frequently been used as topical sunscreen agents, only a few studies in the literature have demonstrated its effectiveness for this specific purpose. Further studies may help to elucidate the protective properties of PABA. Its use as a component of sunscreens has diminished recently because of reports of frequent allergic reactions and cross-sensitivity with other medications.
Pycnogenol®: Pycnogenol® taken by mouth may reduce redness of the skin caused by solar ultraviolet light. Further research is needed before a recommendation can be made. Avoid if allergic or hypersensitive to Pycnogenol®, its components, or members of the Pinaceae family. Use cautiously with diabetes, hypoglycemia, and bleeding disorders. Use cautiously if taking medications that reduce blood cholesterol levels, medications that may increase the risk of bleeding, medications that lower blood pressure, or drugs that affect the immune system. Avoid if pregnant or breastfeeding.
Selenium: Protection from UV damage was initially observed in early research using selenium and other antioxidant supplementation, although there is some evidence that selenium does not prevent light-induced skin redness. Avoid if allergic or sensitive to products containing selenium. Avoid with history of nonmelanoma skin cancer. Selenium is generally regarded as safe for pregnant or breastfeeding women. However, animal research reports that large doses of selenium may lead to birth defects.
Vitamin A: It is unclear whether vitamin A or beta-carotene, taken by mouth or used on the skin with sunscreen, is beneficial in the prevention or treatment of skin cancers or wrinkles. Avoid if allergic or hypersensitive to vitamin A. Vitamin A toxicity can occur if taken at high dosages. Use cautiously with liver disease or alcoholism. Smokers who consume alcohol and beta-carotene may be at increased risk for lung cancer or heart disease. Vitamin A appears safe in pregnant women if taken at recommended doses. However, excess or inadequate vitamin A has been associated with birth defects. Excessive doses of vitamin A have been associated with central nervous system problems. Use cautiously if breastfeeding, because the benefits or dangers to nursing infants have not been clearly established.
Fair negative scientific evidence:
Selenium: Results from the Nutritional Prevention of Cancer (NPC) trial, conducted among 1,312 Americans over a 13-year period, suggest that selenium supplementation given to individuals at high risk of nonmelanoma skin cancer is ineffective in the prevention of basal cell carcinoma and actually increases the risk of squamous cell carcinoma and total nonmelanoma skin cancer. Therefore, selenium supplementation should be avoided in individuals at risk for or with a history of nonmelanoma skin cancer. Avoid if allergic or sensitive to products containing selenium. Avoid with a history of nonmelanoma skin cancer. Selenium is generally regarded as safe for pregnant or breastfeeding women. However, animal research reports that large doses of selenium may lead to birth defects.
Prevention
General: Griscelli syndrome (GS) is inherited, or passed down from family members. Therefore, there is currently no known way to prevent the condition. However, a number of options are available for prospective parents with a family history of GS.
Genetic testing and counseling: Individuals who have GS may meet with a genetic counselor to discuss the risks of having children with the disease. Individuals with a family history of GS may meet with genetic counselors to determine whether they carry any of the various defective genes. Carriers can be determined through detailed family histories or genetic testing.
Known carriers of genes that cause GS may undergo genetic counseling before they conceive a child. Genetic counselors can explain the options and the associated risks of various tests, including preimplantation genetic diagnosis (PGD), amniocentesis, and chorionic villus sampling (CVS).
PGD may be used with in vitro (artificial) fertilization. In PGD, embryos are tested for the defective genes, and only the embryos that are not affected may be implanted. Because GS can be detected in fetus, parents may choose whether to continue the pregnancy. Genetic counselors may assist parents with these difficult decisions.
Author Information
This information has been edited and peer-reviewed by contributors to the Natural Standard Research Collaboration (www.naturalstandard.com).
Bibliography
Natural Standard developed the above evidence-based information based on a thorough systematic review of the available scientific articles. For comprehensive information about alternative and complementary therapies on the professional level, go to www.naturalstandard.com. Selected references are listed below.
Akcakus M, Koklu E, Narin N, et al. Clinical and microscopic hair features of Griscelli syndrome associated with asymmetric crying facies in an infant. Pediatr Dev Pathol. 2007;1. View Abstract
Ashrafi MR, Mohseni M, Yazdani S, et al. Bilateral basal ganglia involvement in a patient with Griscelli syndrome. Eur J Paediatr Neurol. 2006;10(4):207-9. View Abstract
Dinakar C, Lewin S, Kumar KR, et al. Partial albinism, immunodeficiency, hypergammaglobulinemia and Dandy-Walker cyst--a Griscelli syndrome variant. Indian Pediatr. 2003;40(10):1005-8. View Abstract
Griscelli C, Prunieras M. Pigment dilution and immunodeficiency: a new syndrome. Int J Dermatol. 1978;17(10):788-91. View Abstract
Haraldsson A, Weemaes CM, Bakkeren JA, et al. Griscelli disease with cerebral involvement. Eur J Pediatr. 1991;150(6):419-22. View Abstract
Malhotra AK, Bhaskar G, Nanda M, et al. Griscelli syndrome. J Am Acad Dermatol. 2006;55(2):337-40. View Abstract
Mancini AJ, Chan LS, Paller AS. Partial albinism with immunodeficiency: Griscelli syndrome: report of a case and review of the literature. J Am Acad Dermatol. 1998;38(2 Pt 2):295-300. View Abstract
Menasche G, Fischer A, de Saint Basile G. Griscelli syndrome types 1 and 2. Am J Hum Genet. 71: 1237-8, 2002. View Abstract
Menasche G, Pastural E, Feldmann J, et al. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nature Genet. 25: 173-6, 2000. View Abstract
Ménasché G, Ho CH, Sanal O, et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest. 2003 Aug;112(3):450-6. Erratum in: J Clin Invest. 2005 Apr;115(4):1100. View Abstract
Natural Standard: The Authority on Integrative Medicine. www.naturalstandard.com.
Rajadhyax M, Neti G, Crow Y, et al. Neurological presentation of Griscelli syndrome: obstructive hydrocephalus without haematological abnormalities or organomegaly. Brain Dev. 2007;29(4):247-50. View Abstract
Rath S, Jain V, Marwaha RK, et al. Griscelli syndrome. Indian J Pediatr. 2004;71(2):173-5. View Abstract
Smith VV, Anderson G, Malone M, et al. Light microscopic examination of scalp hair samples as an aid in the diagnosis of paediatric disorders: retrospective review of more than 300 cases from a single centre. J Clin Pathol. 2005;58(12):1294-8. View Abstract
Tomita Y, Suzuki T. Genetics of pigmentary disorders. Am J Med Genet C Semin Med Genet. 2004;131C(1):75-81. View Abstract
Copyright © 2013 Natural Standard (www.naturalstandard.com)
The information in this monograph is intended for informational purposes only, and is meant to help users better understand health concerns. Information is based on review of scientific research data, historical practice patterns, and clinical experience. This information should not be interpreted as specific medical advice. Users should consult with a qualified healthcare provider for specific questions regarding therapies, diagnosis and/or health conditions, prior to making therapeutic decisions.
Updated:
March 22, 2017